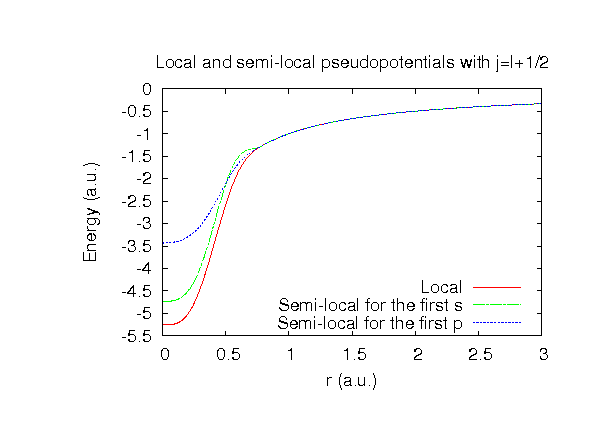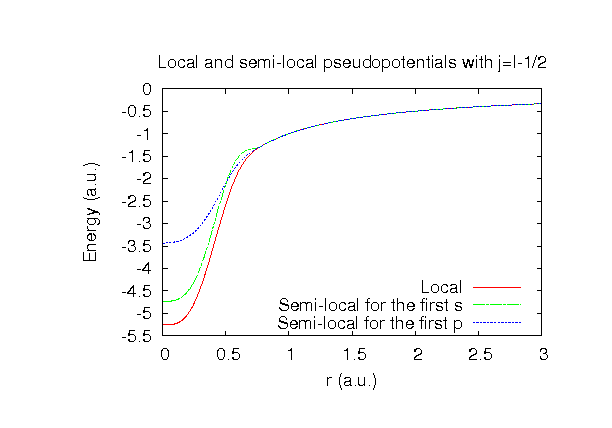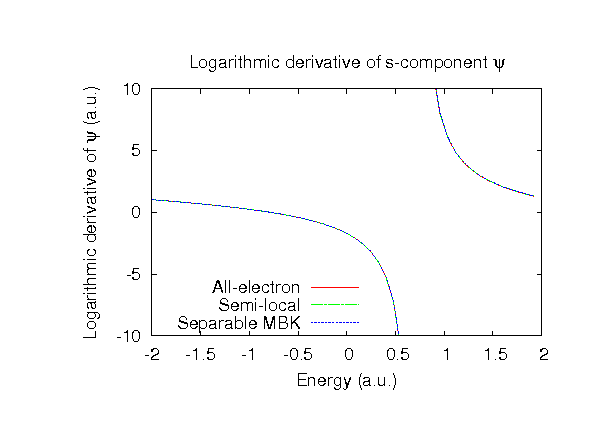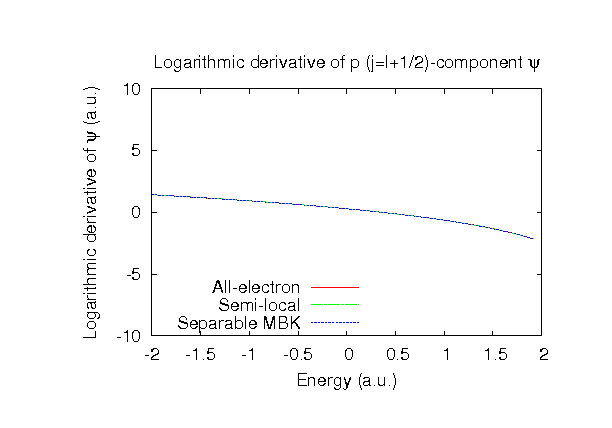
| Equilibrium bond length (Ang.) | Atomization energy (eV) | Atomization energy (couterpoise corrected) (eV) | |
| H7.0-s2p1 | 0.752 | 4.51 | 4.51 |
| H7.0-s3p2 | 0.750 | 4.56 | 4.56 |
| Other calc. | 0.750a | 4.54b | |
| Expt. | 0.741c | 4.75c |
| Equilibrium H-O bond length (Ang.) | Equilibrium H-O-H bond angle (Deg.) | Dipole moment (Debye) | |
| O7.0-s2p2d1, H7.0-s2p1 | 0.971 | 103.7 | 1.82 |
| O7.0-s3p3d2, H7.0-s3p2 | 0.970 | 103.8 | 1.83 |
| Other calc. | 0.969a | 104.2a | 1.81b |
| Expt. | 0.957c | 104.5d | 1.86 c |
| Equilibrium O-O distance (Ang.) | Dipole moment (Debye) | Binding energy (kcal/mol) | Binding energy (couterpoise corrected) (kcal/mol) | |
| O7.0-s2p2d1, H7.0-s2p1 | 2.899 | 2.54 | 5.57 | 5.21 |
| O7.0-s3p3d2, H7.0-s3p2 | 2.897 | 2.45 | 5.48 | 5.48 |
| Other calc. | 2.893a | 5.15 a | ||
| Expt. | 2.98b | 2.60b | 5.44 b |