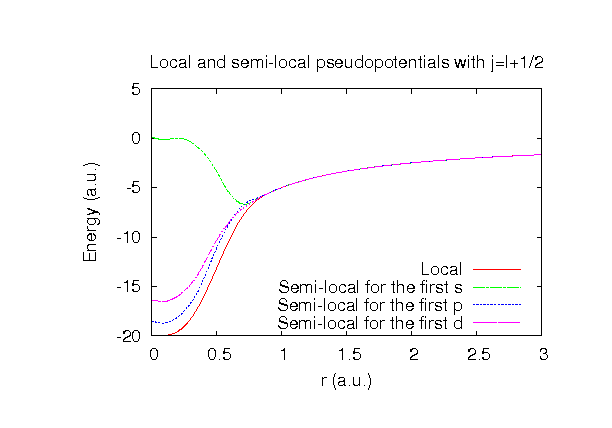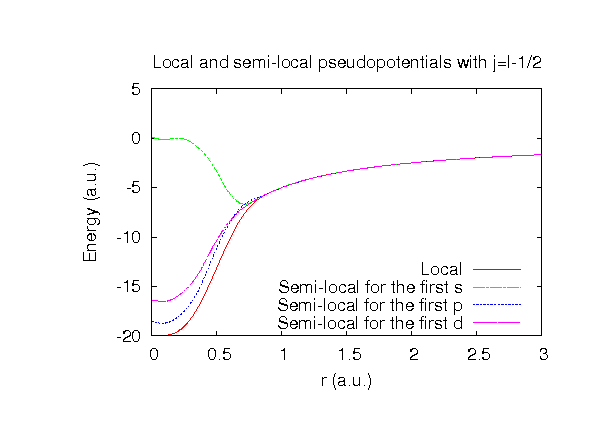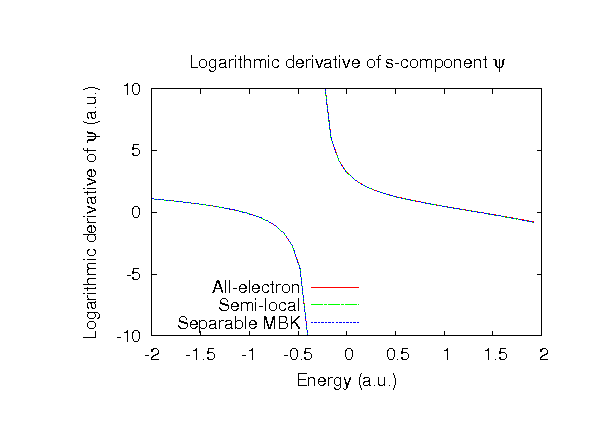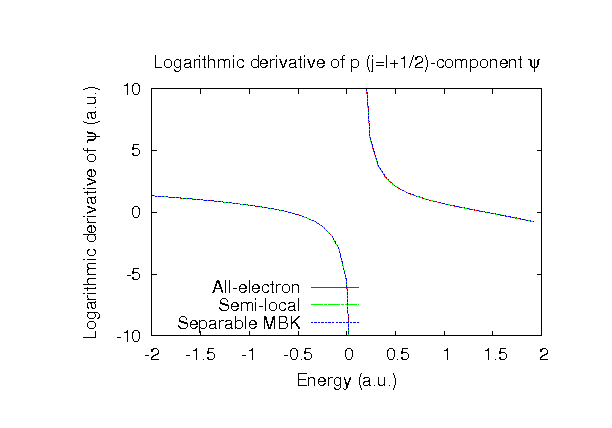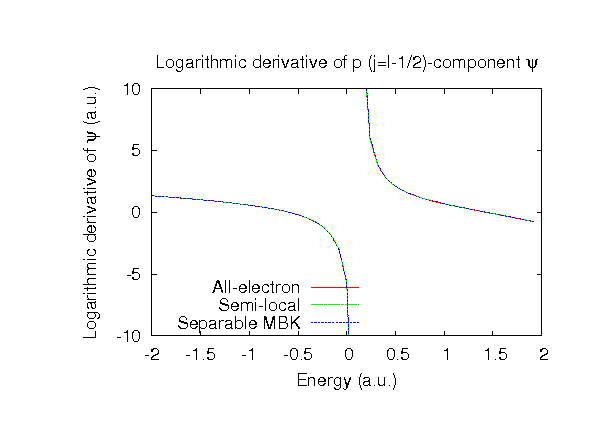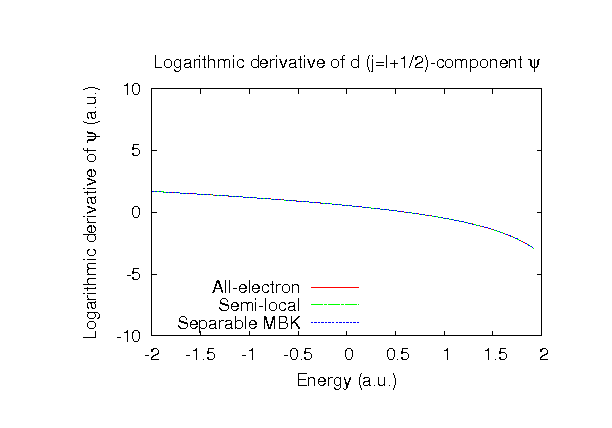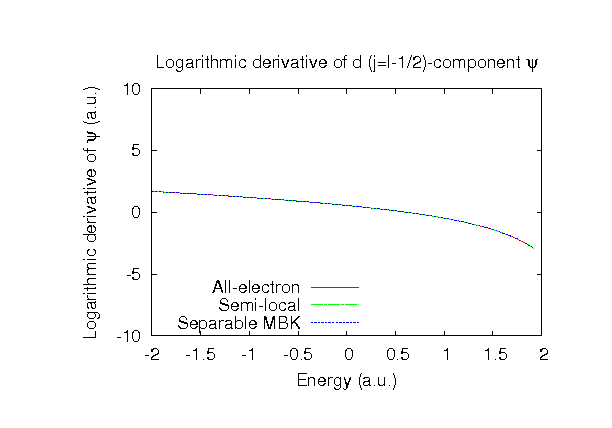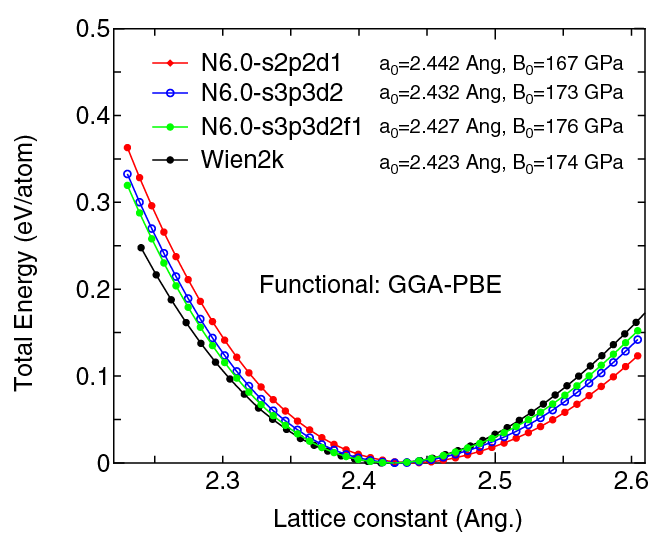
| Equilibrium bond length (Ang.) | Atomization energy (kcal/mol) | Atomization energy (couterpoise corrected) (kcal/mol) | |
| N7.0-s2p2d1 | 1.119 | 221.9 | 221.7 |
| N7.0-s3p3d2 | 1.118 | 223.0 | 222.7 |
| N7.0-s3p3d2f1 | 1.117 | 223.2 | 222.9 |
| Other calc. | 1.111 a | 236.7a | |
| Expt. | 1.098b | 227b |