New Way to Simulate Hydrogen Storage Efficiency of Materials with High Accuracy
Scientists develop new computational method that can simulate hydrogen storage on silicon carbide nanotubes much more accurately than conventional methods
The development of hydrogen technology is invaluable to the future of clean energy, but its progress is hampered by the difficulty in hydrogen storage at room temperature. Computational predictions for storage design go a long way towards reducing the time and cost of development, but how trustworthy are these predictions? Scientists from Japan and USA now create an accurate model to compare with existing methods for the prediction of hydrogen storage on silicon carbide nanotubes.
Hydrogen energy has the potential to be a key measure to meet the United Nations net zero emissions target, but its industrial use has been hindered by the difficulty in its storage and handling. Hydrogen becomes a gas at a very low temperature (-252°C), which makes its storage at room temperature challenging. The interaction between hydrogen and its storage material is simply too weak to persist at room temperature. This makes the design of storage materials crucial to achieving the goal of bringing hydrogen energy into daily use.
This is where computational materials design comes in. A lot of time and effort can be saved during the development of hydrogen technology by designing a material on a computer and simulating its capacity for hydrogen storage. But the predictions become very limited in their use unless they are accurate and can be made at a reasonable computational cost. In a recent study published in ACS Omega, scientists develop a computationally expensive, but highly accurate novel method for predicting hydrogen storage: "Improving prediction reliability for simulations can help accelerate the development of materials for hydrogen fuel storage and lead to a more energy efficient society," says Dr. Kenta Hongo from the Japan Advanced Institute of Science and Technology (JAIST), who led the study.
One of the fundamental forces of attraction between objects is the van der Waals force, which defines the interaction between atoms or molecules based on the distance between them. Since the Van der Waals force is the consequence of quite complicated quantum processes, conventional treatments could not describe it well, and hence the simulations so far are at the level of rough estimations of it. But is it right to do that when simulating hydrogen storage? This was the primary concern of Dr. Hongo and team.
To answer this question, they looked at silicon-carbide nanotubes, one of the most promising materials for hydrogen storage. Using a computational technique called diffusion Monte Carlo (DMC), they created a model that accounted for van der Waals forces when simulating the storage of hydrogen in silicon-carbide nanotubes. Most conventional models consider the interactions between hydrogen and silicon-carbide nanotubes as a whole, but the DMC method uses the power of a supercomputer to reconstruct the interaction mechanism faithfully by following the arrangement of individual electrons. This makes the DMC model the most accurate method of prediction to date. Using the DMC model, the researchers were also able to predict how much energy would be required to dislodge hydrogen from its storage, and how far away the hydrogen was likely to be from the surface of the silicon-carbide nanotube. They then compared the results from their modelling to those obtained via conventional prediction methods.
Conventional prediction methods are usually based on a computational techniques called the density functional theory (DFT). DFT uses functionals (model descriptions of quantum interactions) that describe the spatial variations of electron density to determine the properties of complex systems. While there have been several DFT-based studies on the storage of hydrogen on silicon-carbide nanotubes, none of them have incorporated van der Waals forces in their predictions. Van der Waals-corrected DFT functionals have, however, been employed in the prediction of other materials. Dr. Hongo and team simulated hydrogen storage using a wide range of DFT functionals, those with van der Waals corrections and those without. They found that the DFT functionals without van der Waals corrections misestimated energy required for hydrogen storage by 4-14%. On the other hand, van der Waals-corrected DFT functionals produced results that were quite similar to those of DMC. Moreover, they found that the contribution of the van der Waals force to the storage energy was about 9-29%, which is hardly insignificant.
These findings, Dr. Hongo believes, can be a stepping stone for further innovation in hydrogen storage simulation technology. "Although the DMC method is computationally expensive, it can be used to clarify the peculiarities (tendencies of prediction error) of each prediction method. This will help us understand which prediction to trust, and also how to modify prediction methods to make them more useful," he explains.
With powerful computational predictions like this to speed up its development, it is easy to see hydrogen technology becoming an everyday reality soon!
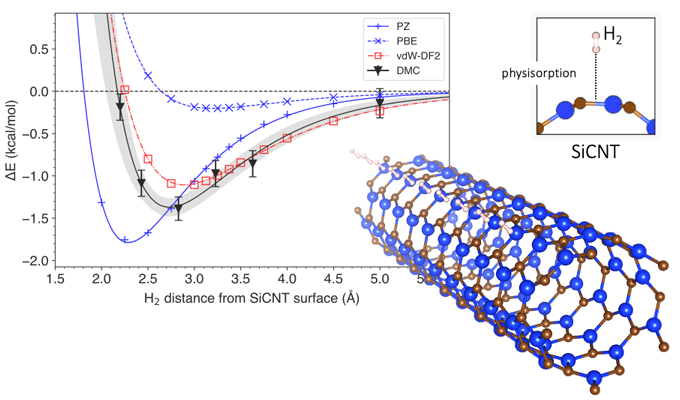
Figure: The energy change associated with hydrogen removal from silicon carbide nanotubes
The graph shows the variation of system energy with the distance of a hydrogen molecule from the surface of a silicon carbide nanotube (bottom right). The depth of the curve signifies the energy required to extract hydrogen from storage. A comparison of prediction methods is presented, with DMC being the most accurate and vdW-DF2 being its closest match.
Figure courtesy: Kenta Hongo from JAIST
Reference
| Title of original paper: | Importance of van der Waals interactions in hydrogen adsorption on a silicon-carbide nanotube revisited with vdW-DFT and quantum Monte Carlo |
| Journal: | ACS Omega |
| DOI: | 10.1021/acsomega.1c03318 |
Funding information
MEXT-KAKENHI (JP19H04692, JP16KK0097, JP16H06439, JP17K17762, JP19K05029, and JP19H05169), FLAGSHIP2020 (project nos. hp190169 and hp190167 at K-computer), the Air Force Office of Scientific Research, JSPS Bilateral Joint Projects (with India DST), and HPCI System Research Project.
September 27 ,2021
