スーパーコンピュータを活用して分子構造シミュレーション上の有名な難問を解決 -直線状シリコン分子の折り曲げ障壁エネルギー値の正確な予測に成功-
スーパーコンピュータを活用して分子構造シミュレーション上の有名な難問を解決
-直線状シリコン分子の折り曲げ障壁エネルギー値の正確な予測に成功-
ポイント
- 分子構造のシミュレーションはよく成功し普及している手法だが、いまだに「シンプルな問題なのにシミュレーションでうまく扱えない」という奇妙な難問がいくつか知られている。
- 直線状シリコン分子を折り曲げるのにどれくらいのエネルギー(折り曲げ障壁エネルギー)が必要になるかのシミュレーション見積もりは、そうした難問の一つである。
- 研究グループはスーパーコンピュータを活用して、第一原理量子モンテカルロ法と呼ばれる超精細な分子シミュレーション手法を適用し、他の手法ではうまく予測できない上記の折り曲げ障壁エネルギー値を正しく予測することに成功した。
【背景と経緯】
目には見えない分子内部の構造をシミュレーションで正しく予測することは、先端科学技術の様々な分野で非常に有用です。例えば、「どの元素が出っぱった外側に来るか」、「最もゆるく結合している元素間の位置はどこか」といったことを知ることによって、化学反応がどのように起こるのか(出っぱった箇所や結合の弱い箇所が攻撃されるのか)、について考えるヒントを得ることできます。このような知見が、新しい物質を合成する方法の開拓につながっていきます。
分子構造のシミュレーションは50年以上の歴史をもち成功と普及を遂げてきましたが、いまだに「シンプルな問題なのにシミュレーションでうまく扱えない」という奇妙な難問がいくつか知られています。その一つが、シリコン分子の折り曲げに関する問題です(図参照)。直線状シリコン分子を折り曲げるのに必要なエネルギー(折り曲げ障壁エネルギー)をシミュレーションで見積もると、実験的に知られている値とは一致しない答が得られます。このこと自体は、さほど驚くことではありません。通常は「よりコストはかかるけど丁寧なシミュレーション」に切り替えていくことで、実験値との整合性はよくなっていきます。
ところが、直線状シリコン分子の場合、分子の状態を記述するパラメータをたくさん設定して、シミュレーションの丁寧な準備を行うほどに余計に整合性が悪くなったり、ある丁寧さのところで急に良くなったりと、系統性が読めない結果になります。これでは「どの程度の精度が欲しいから、どのくらいの丁寧さでシミュレーションしたらいい」という指針が立たず、シミュレーションの結果を信用することができません。直線状シリコン分子の折り曲げ障壁エネルギーの評価は、上記のような理由で、正確な分子シミュレーション法に対する典型的な「挑戦競技」として数多くの研究者が挑戦と失敗を繰り返してきた問題です。
【研究の内容】
北陸先端科学技術大学院大学 環境・エネルギー領域の本郷 研太准教授は、同大学の前園 涼教授、中野 晃佑助教らとともに、「第一原理量子モンテカルロ法」と呼ばれる超精細な分子シミュレーション手法*を専門に、大学の特色であるスーパーコンピュータシステムを活用して研究に取り組んできました。今回、この手法を直線状シリコン分子の折り曲げ障壁エネルギー評価に適用して、実験値を正しく再現する信頼性の高い結果を得ることに成功しました。
従来手法で問題となっていたのは、「記述の丁寧な準備」を段階的に良くしていっても、結果は「段階的に良くなる」というわけではないという点でした。この点に対して、グループが採用したシミュレーション手法では、「準備の丁寧さにはあまり依らずに良好な結果を得る」という特性を示していました。この結果は「シミュレーションの信頼性が、調整パラメータに依存してしまう」、「その調整値で本当によいのか」、といった不安に駆られずに、「シミュレーション結果を信頼できる」という大きな意義を持ちます。
本研究成果は、2022年1月11日(英国時間)に英国王立化学会の科学雑誌「Physical Chemistry Chemical Physics」のオンライン版に掲載されました。
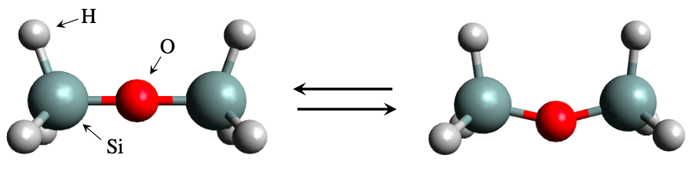
図 直線状シリコン(Si)分子の折り曲げの模式図。現代の分子シミュレーション能力という観点からは、非常にコンパクトな計算規模で扱える分子サイズなのだが、折り曲げに必要なエネルギー(折り曲げ障壁エネルギー値)の予測が非常に難しい奇妙な難問として知られてきた。
【今後の展開】
今回用いた第一原理量子モンテカルロ法は、従来の手法が難渋する「迷宮入りした問題」を解決できる手法であり、その手法の実現にはスーパーコンピュータの利用が必須です。今回の成功事例を皮切りに、スーパーコンピュータの威力を活用して、引き続き、歴史的に知られる分子シミュレーション上の難問解決に挑戦していきます。
【論文情報】
| 掲載誌 | Physical Chemistry Chemical Physics |
| 論文題目 | Diffusion Monte Carlo evaluation of disiloxane linearization barrier(スーパーコンピュータを活用して分子構造シミュレーション上の有名な難問を解決) |
| 著者 | Adie Tri Hanindriyo, Amit Kumar Singh Yadav, Tom Ichibha, Ryo Maezono, Kousuke Nakano, Kenta Hongo |
| 掲載日 | 2022年1月11日(英国時間)にオンライン版に掲載 |
| DOI | 10.1039/d1cp01471d |
【用語説明】
*第一原理量子モンテカルロ法:
「電子レベルのミクロな世界」を記述する方程式を解く方法の一つ。乱数を使って具体的に様々な状況を作り出して「個別の数」の平均をとるアプローチを採用したもの。従来の「関数(数の枠)を使う方法」よりも手間が掛かるが、枠がない分、正確な見積もりを行える。大量の乱数を発生させて数値評価を行うため、スーパーコンピュータの活用が必須となる。
【リンク】
- 研究室 (日/英) [https://www.jaist.ac.jp/is/labs/maezono-lab]
令和4年2月4日
